



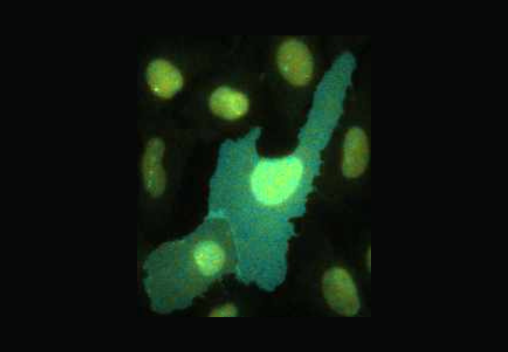
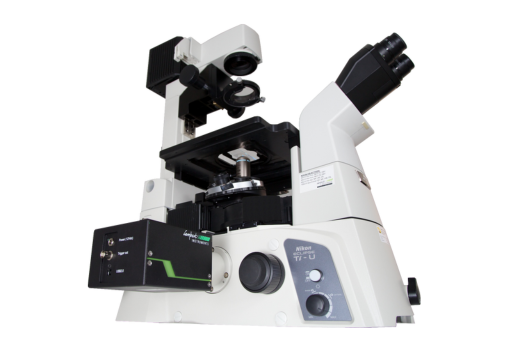
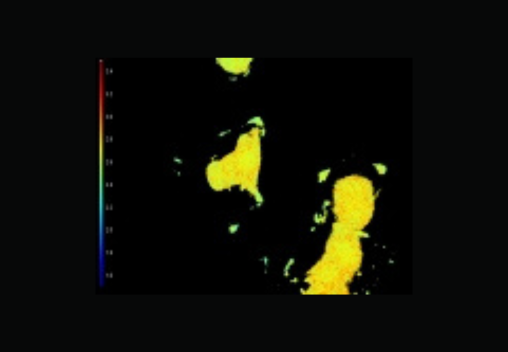
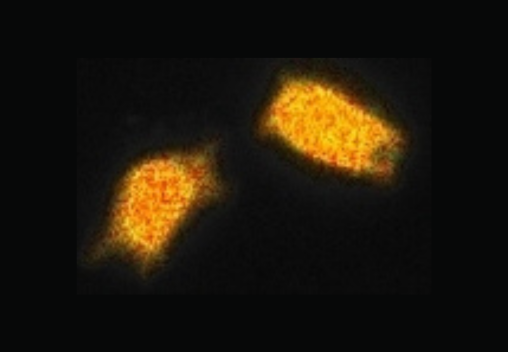

The Lambert Instruments LIFA system is the fastest and easiest way to perform Fluorescence Lifetime Imaging Microscopy (FLIM).
View Product
The TRiCAM is a compact intensified camera. It is designed for scientific and industrial applications that require low-light imaging. With built-in signal generators, the TRiCAM is capable of ultra-short exposures through fast gating and therefore suitable for time-resolved imaging.
View Product
5th floor,
Leonard Springerlaan 19
9727KB Groningen
The Netherlands